Cells are the fundamental unit of tissues and living organisms. Classifying cells into different groups can help researchers understand mechanisms of different biological processes. For instance, research on H3K27M-glioma, a tumor that primarily arises in the midline of the central nervous system in young children, has identified potential therapeutic targets by elucidating its composition of proliferative OPC-like cells sustained by PDGFRA signaling [1].
However, accurately classifying cells is a complex task. Gene expression levels are continuous rather than discrete, following gradient changes. Technical variations in experiments can introduce transcriptional differences that lack biological relevance [2].
The traditional approach relies on annotating cell types based on marker gene expression, but this method can yield conflicting results depending on tissue type, disease state, and experimental design. As a result, researchers must conduct extensive literature reviews and perform significant manual labor to discern real cell types [3]. ML-based approaches have risen in popularity, but differences in reference databases used for model training and ML algorithms can lead to varying annotations, making manual curation unavoidable.
popV: A ML Ensemble for Cell Type annotation
Popular Vote (popV) [4] is a new ML-based tool developed by the Yosef Lab at the Weizmann Institute of Science to address the limitations mentioned above. It employs an ensemble of prediction models with an ontology-based voting scheme to label cells, highlight areas of uncertainty that may require manual scrutiny, and reduce the burden of manual inspection.
Overview of popV’s eight models
popV employs eight machine learning models to transfer labels from an annotated reference dataset, generating predictions alongside a consensus score that reflects confidence in the results. The authors of popV suggest that disagreement across methods often indicates an inaccurate annotation, while agreement among methods typically signals a correct cell type assignment. [4].
The ensemble includes both classical and deep learning-based classifiers, as well as methods that incorporate integration and transfer learning. For example, scANVI is a deep generative model that performs probabilistic label transfer while accounting for uncertainty [5], and OnClass uses ontology-aware classification to assign labels even to unseen cell types [6]. Celltypist is a lightweight logistic regression classifier optimized for speed and interpretability [7]. The remaining classifiers such as SVM [8] and XGBoost [9] are classical supervised learning algorithms known for their strong performance on high-dimensional datasets.
The tool offers three modes for label transfer. Retrain mode trains all classifiers from scratch. Inference mode uses pretrained classifiers to annotate both query and reference cells while constructing a joint embedding with all integration methods. Fast mode utilizes only methods with pretrained classifiers to annotate query cells.
Ground truth data: 718 Hand-Annotated PBMCs Samples from CS Genetics
Peripheral blood mononuclear cells (PBMCs) are widely regarded as the canonical dataset for single-cell RNA-seq analysis. They are easily accessible and relevant to a wide range of scientific questions, including immune responses, disease states, and therapeutic effects. Because of their well-characterized cell type diversity, PBMCs have become the standard for validating new single-cell technologies. In this work, we use PBMC data generated with the CS Genetics SimpleCell™ 3’ Gene Expression assay to evaluate the performance of popV.
718 PBMC samples, processed as 26 experiments, were collected from 16 donors and processed through the CS Genetics SimpleCell™ 3’ Gene Expression assay. The datasets were clustered on a per-experiment basis using Seurat with each cluster manually annotated with a cell type by cell biologists using marker gene expression with individual cells inheriting the annotation of their cluster. This resulted in 24 unique cell types, including a classification of “multiplets” which are assigned when cells had signatures for multiple cell types (5.7% of cells). These manual labels serve as the ground truth for benchmarking model performance. After processing all 26 experiments in Seurat, all cells were combined into a single count matrix of 1,689,880 cells covering 28,340 unique genes.
Cell type labels were mapped to their corresponding Cell Ontology by searching for the manual annotation label using the EMBL-EMI Cell Ontology Lookup Service and selecting the closest match (taking into account relevant markers where appropriate).
Evaluating popV Performance
Here, we compare popV accuracy to cell-biologist-derived annotations of CS Genetics PBMC scRNA-seq data by performing the following analysis:
-
Compare pool-based vs. experiment-based training and testing split strategies.
-
Validate the OnClass model and its ability to propagate unseen cell labels. We chose to highlight the OnClass model specifically because it has the ability to predict cell types that doesn’t exist in the reference, a common challenge for single-cell datasets.
-
Assess label confidence when query datasets contained cell types absent from the reference dataset.
-
Investigate the impact of training dataset size, run mode on model performance.
To evaluate model performance, we calculated accuracies1, weighted accuracies2, and stratified accuracies3 using two different majority voting systems (simple majority voting, popV consensus scoring) and three run modes (retrain, inference, fast).
Pool-based vs. experiment-based splits
To evaluate the performance of the ensemble-based annotation approach, we trained models on subsets of the manually annotated PBMC dataset and assessed their ability to predict cell types in held-out cells. Specifically, we trained models using either 10% (126,435 cells) or 50% (632,175 cells) of the reference dataset and predicted cell types for 39,987 query cells.
To better understand generalizability, we compare two training/testing split strategies while maintaining a similar number of total cells in each setup:
-
Pool-based splitting: A random sample of 50% of all cells was taken and split into 80% training and 20% testing.
-
Experiment-based splitting: 20 (1,264,350 cells) out of 26 experiments were randomly selected for training, with the remaining 6 experiments (425,530 cells) used as unseen query data.
The hypothesis is that experiment-level splitting better simulates true model performance on unseen data and that the accuracy observed with the pool-based approach may be inflated due to test cells coming from the same experiments as the training data. However, similar accuracies were observed across both approaches.
Initial popV Annotation Results
The following results are based on the experiment-level split, using a subset of 39,987 query cells and a training dataset of 126,435 cells.
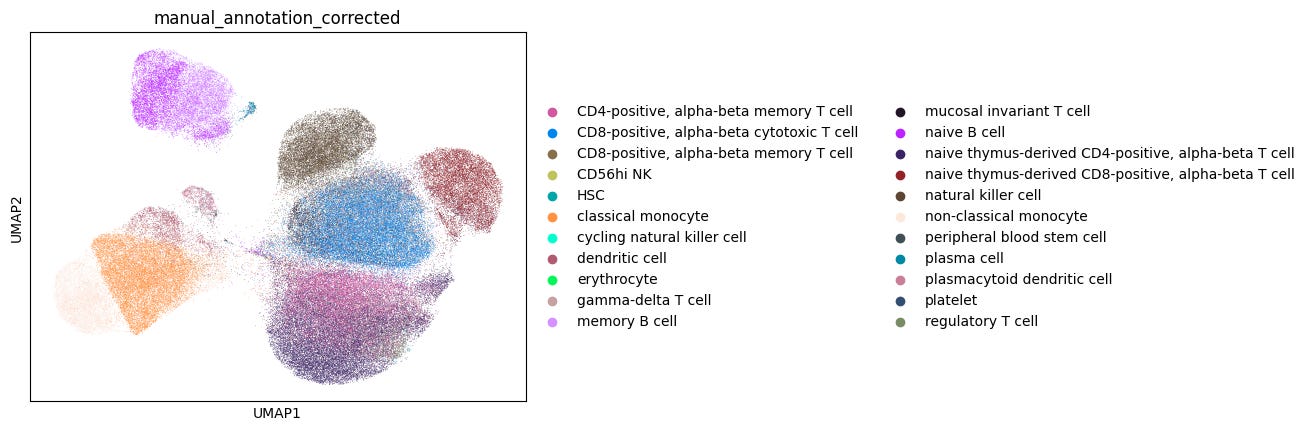
For well-characterized cell types such as classical monocytes, memory B cells, and CD8-positive alpha-beta memory T cells, popV’s ensemble models reached high consensus scores, with nearly all eight models agreeing on their labels.
In contrast, cells located between similar clusters, such as CD8+ αβ cytotoxic and memory T cells, showed low consensus among models, reflecting a challenge in manual annotations that rely on cluster-level markers (Fig. 1). These boundary cells often don’t align cleanly with manual cluster labels, making direct comparisons less meaningful.
This highlights the advantage of cell-level annotation, where each cell is labeled individually rather than assigning a single label to an entire cluster. This approach can yield more accurate results, especially for cells that lie at the boundary between clusters and may express mixed marker profiles.

Ontology-Aware Predictions with OnClass
OnClass is one of the eight models available in popV. It stands out from other models by requiring all reference labels to correspond to valid terms in the Cell Ontology. This promotes standardized, ontology-compliant annotations, a critical advantage for data harmonization across studies. However, this requirement can pose challenges when working with ambiguous or custom labels. In our CS Genetics dataset, some cells were manually annotated as “multiplets” or “proliferating cells”, terms that don’t have corresponding entries in the Cell Ontology and therefore cannot be used during OnClass training.
Despite this challenge, a key strength of OnClass is its ability to predict cell types absent from the reference dataset. It first assigns an annotation from the reference labels (referred to as OnClass_seen) before propagating it to identify a potentially more refined label in the Cell Ontology (even if this label is absent from the reference; referred to as OnClass_prediction), thus generating two different annotations.
When popV includes OnClass in its ensemble, there are two weighting approaches to determine the final annotated labels.
-
Simple majority voting: All model predictions are weighted equally, and only the OnClass_seen label is used.
-
popV consensus scoring: OnClass exerts more influence by contributing both OnClass_seen and OnClass_prediction, with additional weight given based on the path through the ontology hierarchy.
We proceeded to evaluate OnClass performance to decide whether to include OnClass as one of the models contributing to popV’s consensus.
How accurate is OnClass model prediction, and how well can it propagate unseen cell labels?
Assessment of OnClass model prediction
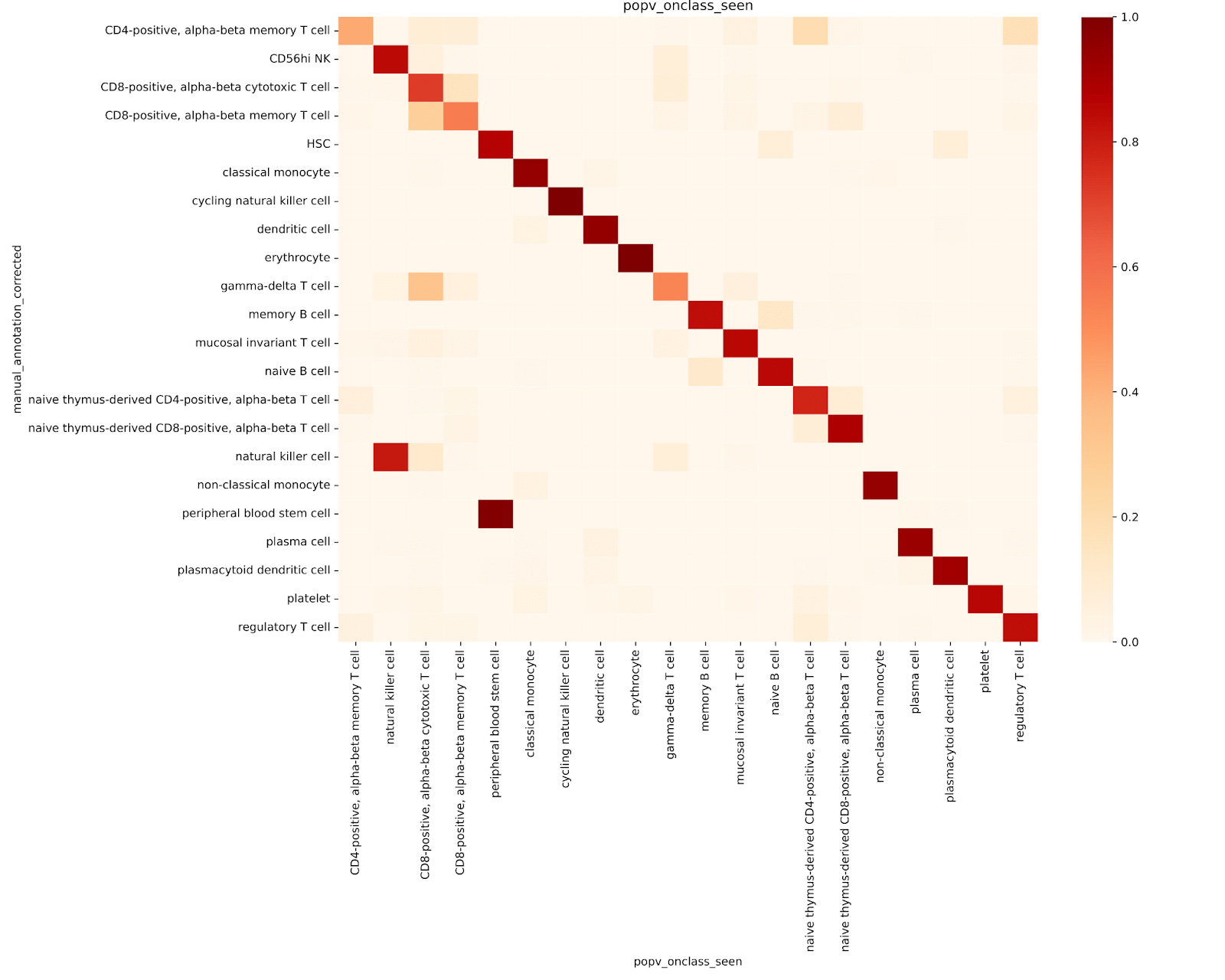
We assessed the accuracy of both the OnClass_seen prediction (i.e. a direct comparison to the manual annotations) and OnClass’s ability to accurately assign cell types through propagation (the OnClass_prediction annotation).
We first evaluated the accuracy of OnClass_seen prediction performance against the ground truth (Fig. 2a). To do this, we ran popV inclusive of the OnClass model, using a 10% downsampling of both the 20 experiment reference and 6 experiment query sets. The OnClass_seen annotations showed a 77% accuracy compared to manual annotations.
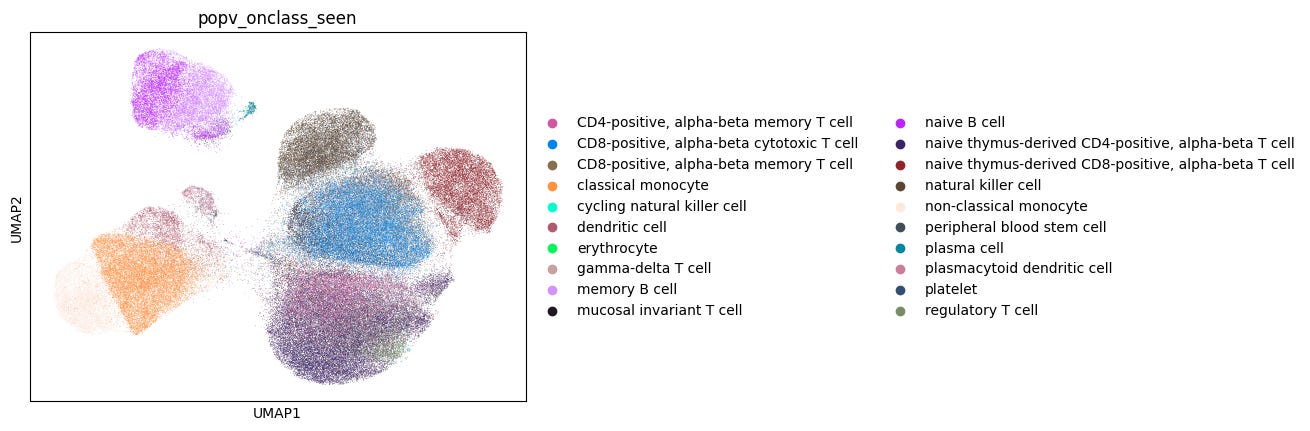
Assessment of OnClass ability to propagate unseen cell labels
To evaluate OnClass’s ability to propagate cell type labels along the ontology hierarchy, we retrained the model using a modified reference dataset. Instead of assigning each cell to its original, fine-grained ontology node, we reassigned each one to its parent node in the ontology tree. This allowed us to test whether OnClass could correctly predict the original, more specific cell type from the broader parent label.
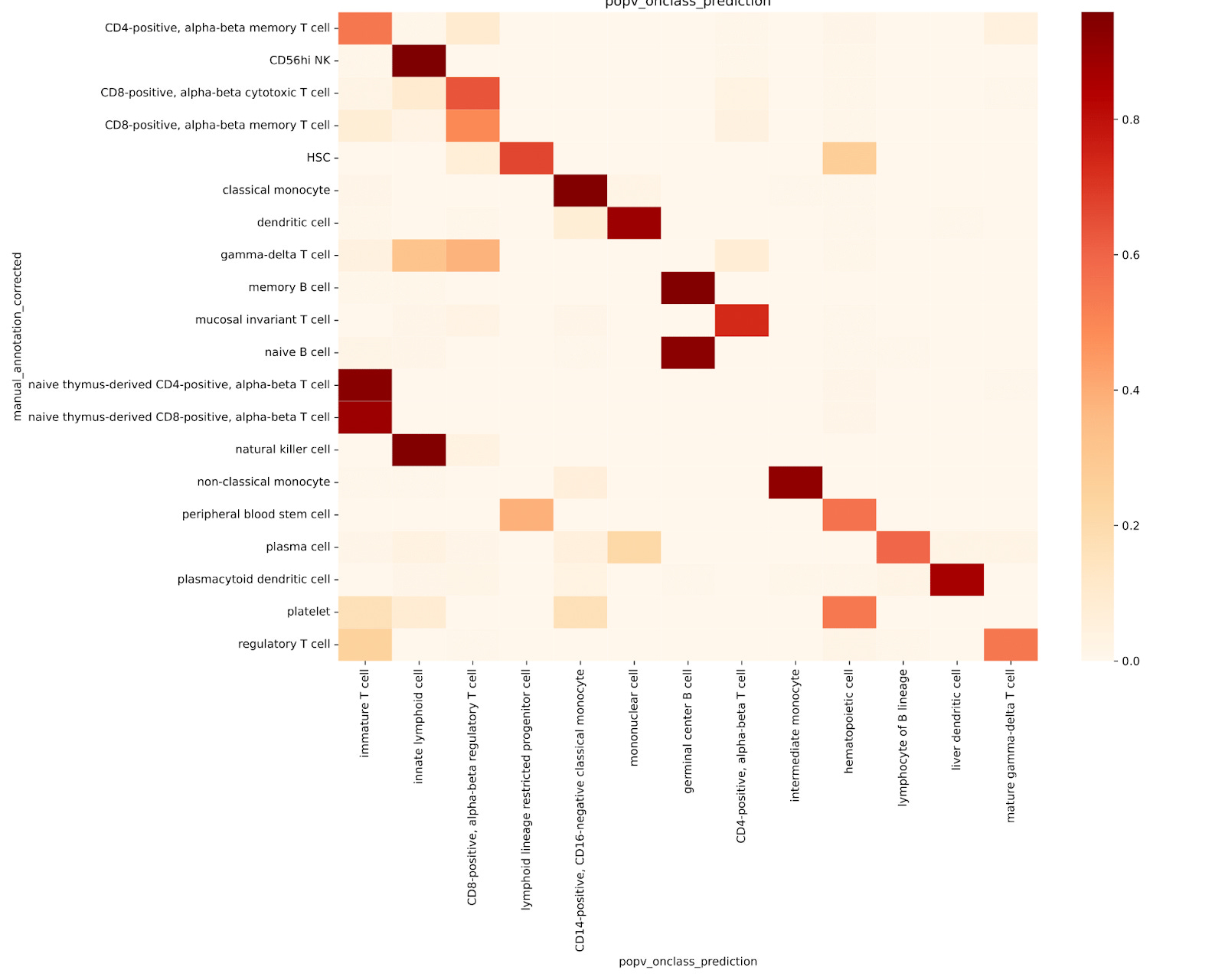
In general, OnClass performed poorly at recovering the original, granular cell types. For example:
-
Most T cells were labeled as immature T cells
-
Monocytes were labeled as intermediate monocytes
-
Mature B cells were often mislabeled as germinal center B cells (Fig. 2b)
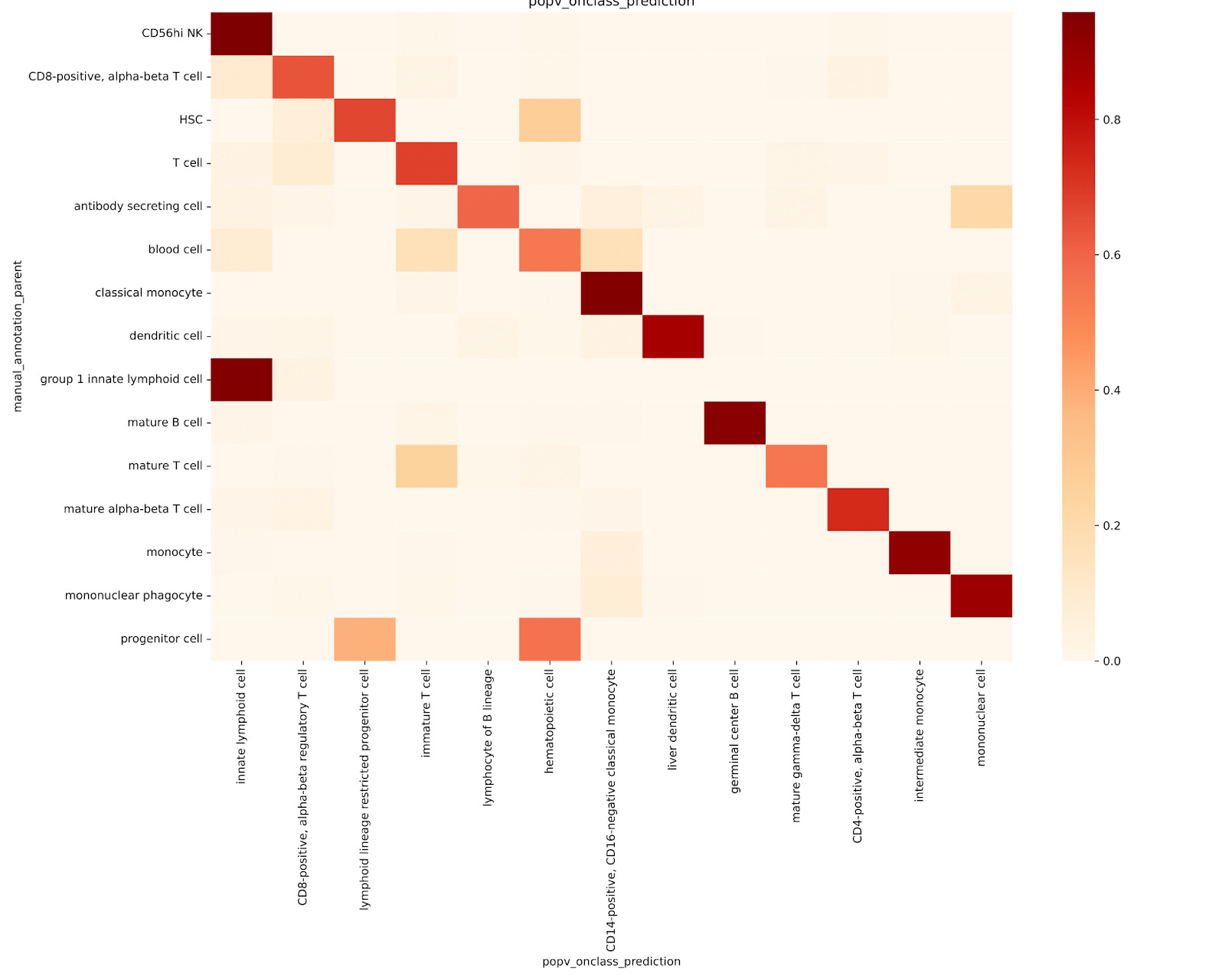
OnClass struggled to identify finer subtypes like memory T cells or naive thymus-derived αβ T cells. There were also clear misclassifications:
-
CD8+ αβ cytotoxic T cells were mislabeled as regulatory T cells
-
Natural killer cells were classified as innate lymphoid cells
-
Platelets were incorrectly labeled as hematopoietic cells, classical monocytes, or even immature T cells (Fig. 2c)
These results underscore the limitations of OnClass’s propagation strategy, particularly in accurately predicting specific or unseen cell types, supporting the authors’ concerns about its reliability in high-resolution annotation tasks.
The Verdict
Despite the poor performance of the propagation approach, OnClass was included as one of the models in popV, given that its OnClass_seen predictions still yielded promising results (Fig. 2d). Consequently, we adopted simple majority voting, rather than consensus scores that also incorporate OnClass_predicted, as the weighting strategy and default setting for cell labeling in the bioinformatics workflow.
How does PopV handle ambiguous cell types?
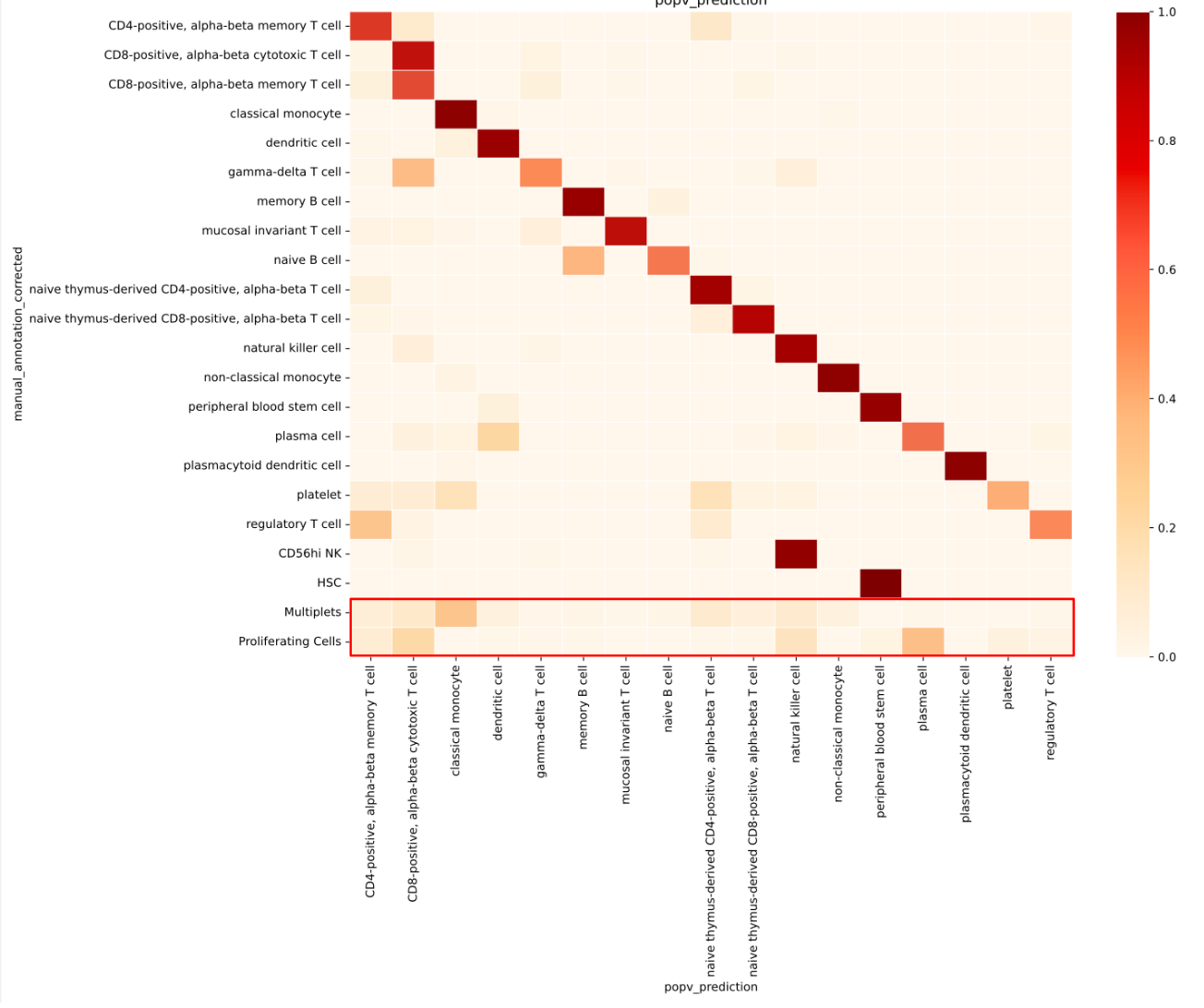
As noted earlier, CS Genetics biologists’ original annotations include ambiguous labels such as “multiplets” and “proliferating cells” when a cluster contains signature markers from multiple different cell types.
To test whether ambiguous labels in the query dataset, such as “multiplets” and “proliferating cells”, would lead to inaccurate annotations, we trained OnClass on a reference dataset that excluded these labels. We then examined how the model annotated the ambiguous cells in the query.
We found that popV assigned a wide range of cell types to these ambiguous cells (Fig. 3), and these assignments were consistently associated with low consensus scores. This low-confidence behavior is valuable: unlike the high-confidence assignments seen for well-defined cell types (Fig. 3), it serves as a built-in signal for users to flag uncertain or mixed-cell cases for further review or exclusion from downstream analysis.
Optimizing popV Parameters
Consensus and Majority Voting yield comparable accuracy
popV supports three modes: retrain, inference, and fast mode, depending on model availability.
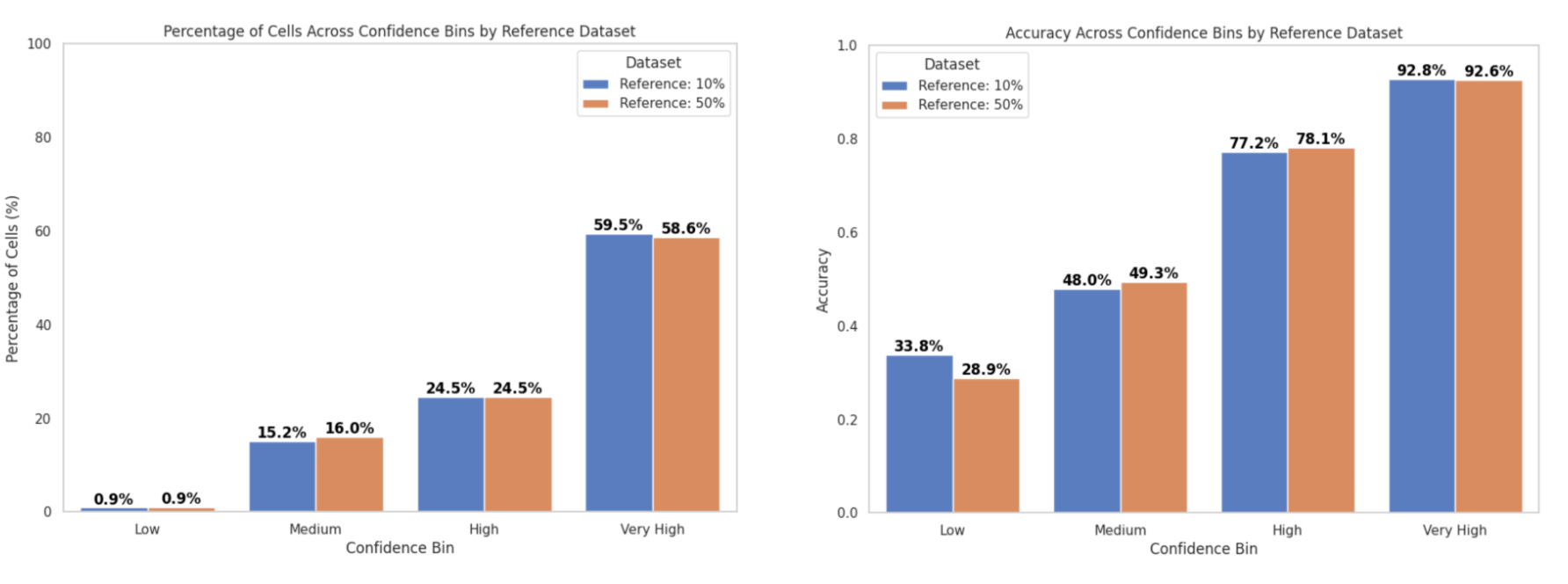
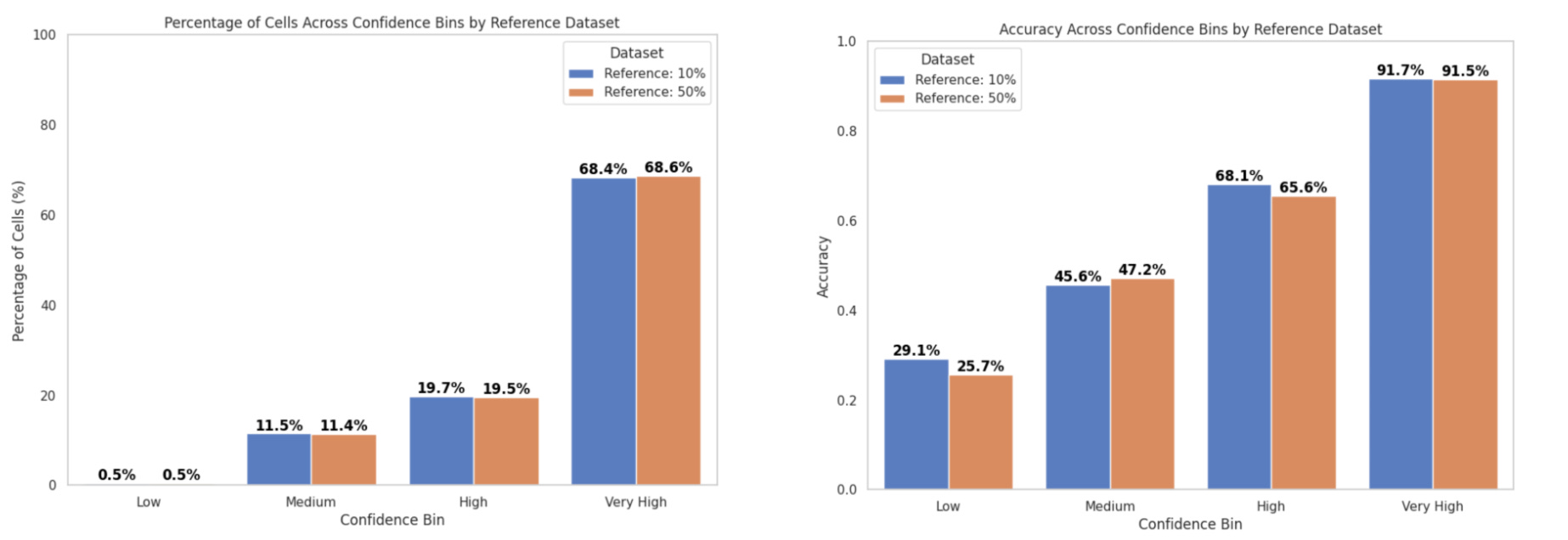
In inference mode, we compared the performance of the consensus score and majority voting for cell type annotation:
-
With the popV consensus score:
-
Accuracy in the Very High confidence bin was ~93%
-
~60% of cells fell into this bin (Fig. 5a)
-
-
With majority voting:
-
Accuracy was ~92%
-
~69% of cells fell into this bin (Fig. 5b)
-
These results are consistent with previous reports from the popV authors, who observed over 95% accuracy when 7 or 8 out of 8 models agreed—criteria that correspond to our Very High confidence bin.
All accuracy values were calculated using 39,987 query cells, excluding ambiguous labels like proliferating cells and multiplets. When including these ambiguous cells (totaling 42,544 cells):
-
Consensus score accuracy dropped slightly to 90%, with 59% of cells in the Very High confidence bin
-
Majority voting had 89% accuracy, with 67% of cells in this bin
Expanding training set size doesn’t improve performance
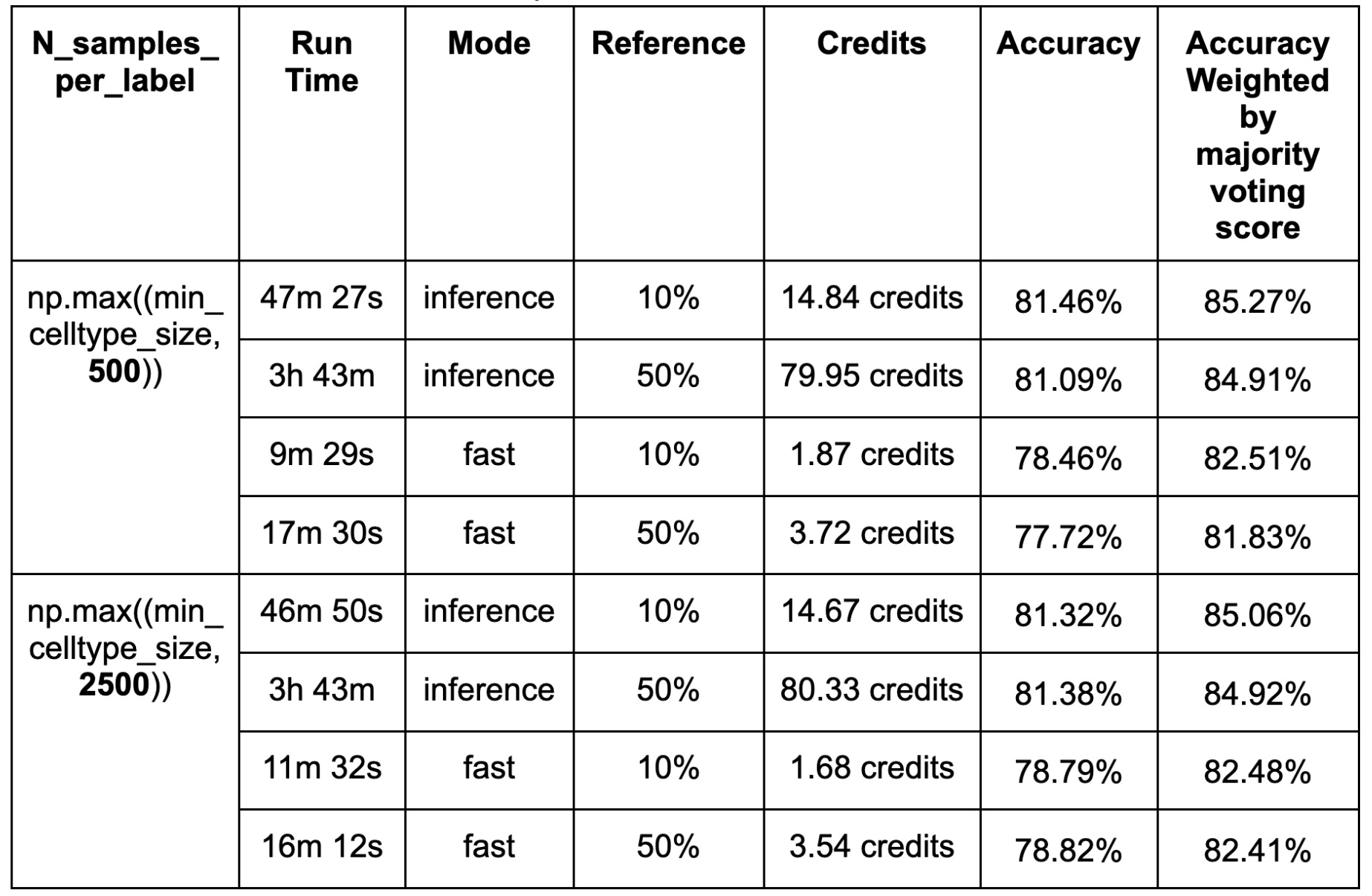
No significant differences in accuracy were observed between models trained on 10% (126,435) and 50% (632,175) of the reference dataset despite a considerable increase in compute time and cost (Table. 1).
This non-significant result may be due to the parameter N_samples_per_label, which controls the number of sampled cells per label, and is set to 500 by default, limiting the impact of increasing the reference pool size. To test this, we increased it to 2500. However, no significant differences were observed. Thus a more likely explanation for the lack of difference between the 10 and 50% training sets is that all cells have relatively similar gene expression profiles, so that increasing the number of cells used for training contributes minimally to performance improvements.
Fast mode offers cost-efficient annotation with minimal accuracy trade-off
Finally, we compared the performance of inference and fast modes in terms of accuracy, computational efficiency, and cost. The inference mode consistently achieved slightly higher accuracy than the fast mode, which is expected given that the fast mode applies a single training epoch in scArches for cell annotation. However, this improvement in accuracy was marginal, with only a ~3% increase when annotating 39,987 cells using 10% of the reference data. In contrast, the inference mode required nearly three times the computational time compared to the fast mode, resulting in significantly higher resource consumption. These findings highlight a trade-off between accuracy and computational efficiency, where the fast mode offers a more cost-effective alternative with only a minimal reduction in accuracy.
a.
b.
The table below summarizes the accuracies, run time and cost between inference and fast mode with 1 L40s GPU. 1 credit is equivalent to $1.
To Conclude
This case study demonstrates that popV confidently annotates most cells accurately while offering mechanisms to identify ambiguous cells requiring further consideration. Given the complexity of cell annotation and the associated expert labor burden, popV, which leverages ensemble ML models and ontology-based consensus voting, can help reduce the manual annotation workload. For cell types that are not present in the training dataset, correctly labeling the cells remains a challenge, but popV mitigates false positives by flagging uncertain cases.
Beyond general annotation, automatic cell typing tools like popV have practical utility in areas like drug discovery, where accurately identifying disease-relevant cell populations and tracking subtle transcriptional changes is crucial for target validation and therapeutic development. Also, in kit and assay validation, popV offers a scalable way to benchmark single-cell assays. By comparing predicted cell types distribution across batches, labs can quickly detect deviations in sample quality.
All eight pretrained models trained on CS Genetics PBMC data, along with the popV workflow, are available on latch.bio, meaning you can start annotating your PBMC data directly on LatchBio.
Get started with popV: Annotate your PBMC single-cell data now on the LatchBio platform. Click here.
For more information on CS Genetics’ SimpleCell technology, please visit csgenetics.com.
Thank you to Imogen West, Ben Hume and Mike Stubbington, who curated CS Genetics PBMC dataset and provided thoughtful review.
Reference
1: https://pubmed.ncbi.nlm.nih.gov/29674595/
2: https://www.csbj.org/article/S2001-0370(21)00019-2/fulltext
3: https://www.sciencedirect.com/science/article/pii/S2001037021000192
4: https://www.nature.com/articles/s41588-024-01993-3
5: https://www.nature.com/articles/s41592-018-0229-2
6: https://pubmed.ncbi.nlm.nih.gov/34548483/
7: https://pmc.ncbi.nlm.nih.gov/articles/PMC7612735/
8: https://dl.acm.org/doi/10.5555/1953048.2078195
9: https://dl.acm.org/doi/10.1145/2939672.2939785
Accuracy refers to correct predictions divided by the total number of predictions.
Weighted accuracy is calculated by weighting the accuracy using the simple majority voting score or popV consensus score, giving more influence to predictions with stronger consensus.
Stratified accuracy evaluates model performance across different confidence levels by grouping cells into confidence bins based on the number of models that agreed on the final predicted label: 1–2 (Low), 3–4 (Medium), 5–6 (High), and 7–8 (Very High).